


Квалификация чистых помещений на фармпроизводстве по GMP | Услуги лаборатории
Для чего нужна аттестация чистых помещений на фармпроизводстве?
В соответствии с «Правилами организации производства и контроля качества лекарственных средств», утвержденными Приказом Минпромторга России от 14.06.2013 N 916, и ГОСТ Р 52249-2009, производство лекарственных средств и фармацевтических субстанций должно вестись в чистых aпомещениях.
При производстве лекарственных средств особое значение имеет понятие «стерильность», означающие «отсутствие живых микроорганизмов». Для обеспечения стерильности на фармпроизводстве технологические операции при производстве лекарственных препаратов, как проходящих финишную стерилизацию, так и производимых в асептических условиях, должны производиться в чистых помещениях или чистых зонах.
Основные требования к чистым помещениям на фармацевтическом производстве
Требования к чистым помещениям для асептического фармпроизводства и производства лекарственных средств, которые могут быть подвергнуты финишной стерилизации, отличаются. Технологический процесс розлива / наполнения (критический процесс) при производстве лекарственных средств, не подвергаемых финишной стерилизации в упаковке, требует чистой зоны «А», окруженной чистой зоной класса «В», чтобы свести к минимум риск контаминации готовой продукции частицами и микроорганизмами.
Для фармацевтической продукции, которая проходит финишную стерилизацию, «Правилами организации производства и контроля качества лекарственных средств» установлены менее жесткие требования, в частности, наполнение продуктами, подлежащими финишной стерилизации, может проводиться в производственной среде класса С, однако, при повышенном риске контаминации (если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации), наполнение так же должно проводиться в чистой зоне класса А (но с окружающей средой, по крайней мере, класса С).
Требования к оснащенному и эксплуатируемому состоянию должны быть установлены для каждого чистого помещения или комплекса чистых помещений.
| Асептическое производство | |
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Асептическое приготовление и наполнение |
| В |
Зоны, окружающие зону «А» |
| С |
Приготовление растворов для фильтрации |
| D |
Операции с материалами после мойки |
|
Операции с продукцией, подлежащей финишной стерилизации |
|
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Операции с продуктом, когда его нельзя подвергать риску загрязнения |
| В |
Зоны, окружающие зону «А» |
| C |
Наполнение продуктом |
| D |
Приготовление растворов и подготовка первичной упаковки, материалов и др. для последующего наполнения |
Классы чистоты помещений в фармацевтическом производстве
ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» определяет типы чистых зон (А, B, С, D) и соответствующие им классы чистоты по ИСО (ГОСТ Р ИСО 14644-1-2017) для различных чистых помещений фармацевтического производства и отдельных технологических процессов.
| Тип чистой зоны | Максимально допустимое число частиц в 1 м3 воздуха при размере частиц, равном или большем | |||
|---|---|---|---|---|
| В оснащенном состоянии | В эксплуатируемом состоянии | |||
| 0,5 мкм | 5,0 мкм | 0,5 мкм | 5,0 мкм | |
| А | 3520 | 20 | 3520 | |
| В | 3520 | 29 | 352000 | 2900 |
| С | 352000 | 2900 | 3520000 | 29000 |
| D | 3520000 | 29000 | – | – |
Как проводится валидация чистых помещений на фарме?
Основным параметром чистого помещения, требующим проверки при проведении аттестации на фармацевтическом производстве, является класс чистоты помещения по ИСО (тип чистой зоны).
Для чистых зон класса А (ИСО 4.8 по частицам с размерами > 5,0 мкм и ИСО 5 по частицам с размерами >0,5 мкм) допустимые концентрации частиц составляют 20 и 3520 шт/м3 соответственно. Для измерения низких концентраций частиц с размерами >5 мкм отбирается не менее 1 м3 воздуха. В ряде случаев может использоваться метод последовательного счета, позволяющий ускорить процесс анализа без ущерба для точности результатов. В частности, этот метод применяется при проверке ламинарных боксов.
В зависимости от особенностей конкретного фармацевтического производства и технологического процесса, проводится также проверка параметров микроклимата (температура и влажность, стабильность поддержания параметров микроклимата), измерение расхода приточного и вытяжного воздуха и кратности воздухообмена и других необходимых параметров.
При наличии в чистой зоне класса «А» однонаправленного потока воздуха, измеряется скорость воздушного потока и оценивается его равномерность. Скорость однонаправленного потока в соответствии с правилами GMP должна лежать в пределах 0,36-0,54 м/с. В закрытых изолирующих устройствах и ламинарных боксах допустим однонаправленный поток воздуха с меньшей скоростью, при этом проверка проводится на соответствие скорости и расхода воздуха технической документации (проектной документации или паспорту бокса).
При аттестации в оснащенном и эксплуатируемом состоянии может также проводиться визуализация воздушных потоков вблизи оборудования, показывающая влияние выступающих частей оборудования на движение воздуха и позволяющая оценить риск возникновения застойных зон.
Еще одним важным показателем, требующим проверки, является целостность финишных HEPA-фильтров. При наличии утечек в самих фильтрах или их уплотнении достижение помещением требуемого класса чистоты может быть затруднено, риск загрязнения продукции резко возрастает.
Испытание фильтров на утечку проводится с использованием генератора аэрозольных частиц и позволяет локализовать утечку и заменить или отремонтировать поврежденные фильтры. При проведении работ по проверке HEPA-фильтров в ходе аттестации чистых помещений на фармпроизводстве специалисты лаборатории Академлаб сразу же информируют технолога и менеджера по качеству фармпроизводства о выявленных утечках, что позволяет заменить или отремонтировать фильтры на месте и сразу провести повторные испытания, что экономит время и средства.
Для чего привлекать к аттестации чистых помещений стороннюю аккредитованную лабораторию?
При наличии у собственной службы качества всего необходимого оборудования для проведения мониторинга и аттестации чистых помещений, привлечение сторонней лаборатории дает дополнительные преимущества и массу новой информации:
- Возможность проведения сравнительных испытаний
- Подтверждение компетентности собственной службы качества
- Наличие протоколов независимой аккредитованной лаборатории, подтверждающих соответствие чистых помещений требования GMP, снимающих множество вопросов в ходе внешних инспекций
Кроме того, фармацевтическая система качества по ГОСТ Р 52537-2006 «Производство лекарственных средств. Система обеспечения качества. Общие требования» регламентирует проведение внутренних аудитов с привлечением независимых лабораторий:
Аудит может выполняться <…> с привлечением, при необходимости, аккредитованных Испытательных лабораторий для проверки соответствия установленным требованиям (аттестации) оборудования, чистых помещений и процессов.
Отчет об испытаниях чистых помещений на фармацевтическом производстве
Отчет о проведенных испытаниях чистых помещений фармацевтического предприятия включает в себя протоколы измерений и информацию о соответствии помещений требованиям GMP и проектной документации, а также, при необходимости, дополнительную информацию — первичные данные измерений, данные визуализации потоков и т.п.
Отправить заявку
Квалификация чистых помещений | Центр Валидации
Чистое помещение – это техническое решение для определенного помещения, которое позволяет поддерживать концентрацию частиц загрязняющих веществ в определенных пределах в соответствии с требованиями стандартов производства различных продуктов. С целью подтверждения функционирования согласно всем нормам и требованиям проводится квалификация чистых помещений.
Квалификация чистых помещений – это процесс документального подтверждения соответствия чистого помещения (чистой зоны) заданному классу чистоты.
Во время квалификации чистых помещений может проводиться:
-
Контроль монтажа помещений и оборудования СВиК;
-
Квалификация систем вентиляции и кондиционирования;
-
Тестирование НЕРА фильтров;
-
Настройка и балансировка систем вентиляции;
-
Мониторинг чистых помещений;
-
Квалификация ламинарных шкафов;
-
Квалификация ламинарных боксов;
-
Квалификация шкафов биологической безопасности и биологической защиты;
-
Финальная аттестация чистых помещений.
По согласованию с Заказчиком может быть проведена балансировка (настройка) системы вентиляции и обучение персонала по безопасной замене фильтров (для биолабораторий).
Мы проводим все стадии квалификации чистых помещений, ламинарных боксов, боксов биологической защиты, зон отбора проб и др.:
-
DQ (Design qualification) — квалификация проекта;
-
IQ (I nstallation qualification) — квалификация монтажа;
-
OQ (Operation qualification) — квалификация функционирования;
-
PQ (Performance qualification) — квалификация эксплуатации.
Наш опыт по квалификации чистых помещений:
-
Участки производства стерильных и нестерильных лекарственных средств;
-
Участки производства иммунобиологических препаратов, вакцин;
-
Микробиологические лаборатории;
-
Родильные отделения;
-
Микроэлектроника;
-
Лаборатории биобезопасности III класса;
-
Центры гематоонкологии.
Наш опыт по квалификации чистых помещений позволяет провести все тесты в максимально сжатые сроки, чтобы минимизировать длительность остановки производственного процесса для проведения квалификации чистых помещений.
Зачем проводить?
 В соответствии с требованиями GMP, производство лекарственных средств должно осуществляться в чистых помещениях, оборудованных соответствующими Системами Обогрева, Вентиляции и Кондиционирования (HVAC — Heating Ventilation and Air Conditioning Systems). Смысл такого требования заключается в создании чистой окружающей среды для производства, обеспечении защиты для критических технологических операций и предотвращения риска для качества продукции. Воздух в чистых помещениях непосредственно влияет на качество выпускаемой продукции, системы HVAC и все чистые помещения относятся к критическим системам и, соответственно, подлежат квалификации.
В соответствии с требованиями GMP, производство лекарственных средств должно осуществляться в чистых помещениях, оборудованных соответствующими Системами Обогрева, Вентиляции и Кондиционирования (HVAC — Heating Ventilation and Air Conditioning Systems). Смысл такого требования заключается в создании чистой окружающей среды для производства, обеспечении защиты для критических технологических операций и предотвращения риска для качества продукции. Воздух в чистых помещениях непосредственно влияет на качество выпускаемой продукции, системы HVAC и все чистые помещения относятся к критическим системам и, соответственно, подлежат квалификации.
Системы HVAC и чистые помещения, кроме того, относятся к системам с непрерывным режимом работы, что обычно требует проведения для таких систем регулярного мониторинга критических функций и параметров, а также реквалификации на периодической основе.
Все необходимые тесты норм чистоты помещений в максимально короткие сроки
Квалификация чистых помещений на фармпроизводстве по GMP | Услуги лаборатории
Для чего нужна аттестация чистых помещений на фармпроизводстве?
В соответствии с «Правилами организации производства и контроля качества лекарственных средств», утвержденными Приказом Минпромторга России от 14.06.2013 N 916, и ГОСТ Р 52249-2009, производство лекарственных средств и фармацевтических субстанций должно вестись в чистых aпомещениях.
При производстве лекарственных средств особое значение имеет понятие «стерильность», означающие «отсутствие живых микроорганизмов». Для обеспечения стерильности на фармпроизводстве технологические операции при производстве лекарственных препаратов, как проходящих финишную стерилизацию, так и производимых в асептических условиях, должны производиться в чистых помещениях или чистых зонах.
Основные требования к чистым помещениям на фармацевтическом производстве
Требования к чистым помещениям для асептического фармпроизводства и производства лекарственных средств, которые могут быть подвергнуты финишной стерилизации, отличаются. Технологический процесс розлива / наполнения (критический процесс) при производстве лекарственных средств, не подвергаемых финишной стерилизации в упаковке, требует чистой зоны «А», окруженной чистой зоной класса «В», чтобы свести к минимум риск контаминации готовой продукции частицами и микроорганизмами.
Для фармацевтической продукции, которая проходит финишную стерилизацию, «Правилами организации производства и контроля качества лекарственных средств» установлены менее жесткие требования, в частности, наполнение продуктами, подлежащими финишной стерилизации, может проводиться в производственной среде класса С, однако, при повышенном риске контаминации (если операции наполнения проходят медленно или упаковки имеют широкое горло, или их необходимо держать открытыми более нескольких секунд до герметизации), наполнение так же должно проводиться в чистой зоне класса А (но с окружающей средой, по крайней мере, класса С).
Требования к оснащенному и эксплуатируемому состоянию должны быть установлены для каждого чистого помещения или комплекса чистых помещений.
| Асептическое производство | |
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Асептическое приготовление и наполнение |
| В |
Зоны, окружающие зону «А» |
| С |
Приготовление растворов для фильтрации |
| D |
Операции с материалами после мойки |
| Операции с продукцией, подлежащей финишной стерилизации | |
|---|---|
| Тип зоны | Выполняемые операции |
| А |
Операции с продуктом, когда его нельзя подвергать риску загрязнения |
| В |
Зоны, окружающие зону «А» |
| C |
Наполнение продуктом |
| D |
Приготовление растворов и подготовка первичной упаковки, материалов и др. для последующего наполнения |
Классы чистоты помещений в фармацевтическом производстве
ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» определяет типы чистых зон (А, B, С, D) и соответствующие им классы чистоты по ИСО (ГОСТ Р ИСО 14644-1-2017) для различных чистых помещений фармацевтического производства и отдельных технологических процессов.
| Тип чистой зоны | Максимально допустимое число частиц в 1 м3 воздуха при размере частиц, равном или большем | |||
|---|---|---|---|---|
| В оснащенном состоянии | В эксплуатируемом состоянии | |||
| 0,5 мкм | 5,0 мкм | 0,5 мкм | 5,0 мкм | |
| А | 3520 | 20 | 3520 | 20 |
| В | 3520 | 29 | 352000 | 2900 |
| С | 352000 | 2900 | 3520000 | 29000 |
| D | 3520000 | 29000 | – | – |
Как проводится валидация чистых помещений на фарме?
Основным параметром чистого помещения, требующим проверки при проведении аттестации на фармацевтическом производстве, является класс чистоты помещения по ИСО (тип чистой зоны).
Для чистых зон класса А (ИСО 4.8 по частицам с размерами > 5,0 мкм и ИСО 5 по частицам с размерами >0,5 мкм) допустимые концентрации частиц составляют 20 и 3520 шт/м3 соответственно. Для измерения низких концентраций частиц с размерами >5 мкм отбирается не менее 1 м3 воздуха. В ряде случаев может использоваться метод последовательного счета, позволяющий ускорить процесс анализа без ущерба для точности результатов. В частности, этот метод применяется при проверке ламинарных боксов.
В зависимости от особенностей конкретного фармацевтического производства и технологического процесса, проводится также проверка параметров микроклимата (температура и влажность, стабильность поддержания параметров микроклимата), измерение расхода приточного и вытяжного воздуха и кратности воздухообмена и других необходимых параметров.
При наличии в чистой зоне класса «А» однонаправленного потока воздуха, измеряется скорость воздушного потока и оценивается его равномерность. Скорость однонаправленного потока в соответствии с правилами GMP должна лежать в пределах 0,36-0,54 м/с. В закрытых изолирующих устройствах и ламинарных боксах допустим однонаправленный поток воздуха с меньшей скоростью, при этом проверка проводится на соответствие скорости и расхода воздуха технической документации (проектной документации или паспорту бокса).
При аттестации в оснащенном и эксплуатируемом состоянии может также проводиться визуализация воздушных потоков вблизи оборудования, показывающая влияние выступающих частей оборудования на движение воздуха и позволяющая оценить риск возникновения застойных зон.
Еще одним важным показателем, требующим проверки, является целостность финишных HEPA-фильтров. При наличии утечек в самих фильтрах или их уплотнении достижение помещением требуемого класса чистоты может быть затруднено, риск загрязнения продукции резко возрастает.
Испытание фильтров на утечку проводится с использованием генератора аэрозольных частиц и позволяет локализовать утечку и заменить или отремонтировать поврежденные фильтры. При проведении работ по проверке HEPA-фильтров в ходе аттестации чистых помещений на фармпроизводстве специалисты лаборатории Академлаб сразу же информируют технолога и менеджера по качеству фармпроизводства о выявленных утечках, что позволяет заменить или отремонтировать фильтры на месте и сразу провести повторные испытания, что экономит время и средства.
Для чего привлекать к аттестации чистых помещений стороннюю аккредитованную лабораторию?
При наличии у собственной службы качества всего необходимого оборудования для проведения мониторинга и аттестации чистых помещений, привлечение сторонней лаборатории дает дополнительные преимущества и массу новой информации:
- Возможность проведения сравнительных испытаний
- Подтверждение компетентности собственной службы качества
- Наличие протоколов независимой аккредитованной лаборатории, подтверждающих соответствие чистых помещений требования GMP, снимающих множество вопросов в ходе внешних инспекций
Кроме того, фармацевтическая система качества по ГОСТ Р 52537-2006 «Производство лекарственных средств. Система обеспечения качества. Общие требования» регламентирует проведение внутренних аудитов с привлечением независимых лабораторий:
Аудит может выполняться <…> с привлечением, при необходимости, аккредитованных Испытательных лабораторий для проверки соответствия установленным требованиям (аттестации) оборудования, чистых помещений и процессов.
Отчет об испытаниях чистых помещений на фармацевтическом производстве
Отчет о проведенных испытаниях чистых помещений фармацевтического предприятия включает в себя протоколы измерений и информацию о соответствии помещений требованиям GMP и проектной документации, а также, при необходимости, дополнительную информацию — первичные данные измерений, данные визуализации потоков и т.п.
Отправить заявку
Категории помещений по электробезопасности: классификация
Вопросы электробезопасности на производстве, это лишь часть всех мероприятий и требований промышленной безопасности. Выполнение требований многочисленных инструкций и правил, обеспечение безопасных условий труда возложено кодексом РФ о труде, на работодателя. Он же несёт ответственность. Мера её зависит от тяжести последствий нарушений или невыполнения требований промбезопасности и охраны труда. Руководитель предприятия особенно если оно большое назначает работников ответственных за выполнение требований ОТ, пожарной и электробезопасности.
Нормативные документы
Правила устройства электроустановок (ПУЭ) распространяются на все строящиеся и реконструируемые электроустановки переменного и постоянного тока напряжением 750 кВ и обязательны к исполнению, независимо от отраслевой принадлежности и формы собственности. Новое, 7 издание постоянно пополняется по мере переработки материалов и их согласования с заинтересованными ведомствами и утверждения в министерстве.
Тем не менее эксперты утверждают, что издание не охватывает всего объёма необходимых изменений, и вероятно в недалёком будущем в свет выйдет очередное дополненное издание ПУЭ.
Требования по охране труда и пожарной безопасности при работе с электроустановками тоже изменяются.
Категории помещений по электробезопасности, ПУЭ
В соответствии с правилами устройства — электроустановки это совокупность технологического электрооборудования машин и аппаратов вместе с сооружениями в которых они установлены предназначенные для выработки или передачи, трансформации и перераспределения, преобразования в другие виды энергии. Делятся на:
- Наружные (открытые). Расположенные на открытой местности не защищённые от атмосферных воздействий.
- Закрытые (внутренние). Находящиеся в зданиях защищающих их от атмосферных воздействий.
Электропомещения — различные сооружения, здания или отгороженные части помещения в котором расположено электрооборудование с доступом только для квалифицированного персонала занятого обслуживанием. Все эти помещения по электробезопасности подразделяются на 4 категории:
- Помещения без повышенной опасности.
- Помещения с повышенной опасностью.
- Особо опасные.
- Территории, на которых установлены открытые электроустановки, где возможно поражение людей током, относятся к особо опасным помещениям.
1 категория
В ПУЭ говорится, что это помещения, в которых нет условий для возникновения повышенной или особой опасности. Что это за помещения?
Помещения без повышенной опасности это обычные жилые или офисные здания. Предприятия социальной сферы детские дошкольные учреждения, школы, больницы и так далее. Основные требования для 1-й категории:
- Сухие — 60% и влажные помещения влажность воздуха в которых не должна превышать 75%.
- Работающая приточно-вытяжная вентиляция. Не должно быть токопроводящей пыли и химических соединений в воздухе.
- Температура окружающего воздуха не превышает +35°C.
- Покрытие пола должно быть выполнено из материалов не проводящих электричество.


В эту категорию можно отнести и некоторые производства и цеха, но вышеперечисленные условия должны быть соблюдены. Требования по охране труда к работникам ограничиваются вводным инструктажем, и с периодичностью два раза в год инструктажем на рабочем месте.
К обслуживанию электроустановок допускаются специалисты с 3-й группой допуска до 1000 вольт. Ответственный, за электрохозяйство назначается из состава ИТР с 4-й группой.
2 категория: опасные помещения по электробезопасности
Действующая по настоящее время классификация помещений по электробезопасности ПУЭ ко второй категории относит отвечающие следующим критериям:
- Сырые помещения. Влажность более 75%
- В воздухе возможно наличие токопроводящей пыли.
- Цеха с высоким содержанием в воздухе химических соединений.
- Полы выполнены из материалов способных, проводить электричество (металл, земля, железобетон, кирпич и пр.).
- Помещения с высокой температурой.
- Возможность, одновременно прикоснуться к станку или другому оборудованию с одной стороны и металлическим частям (корпусам) электрооборудования или открытым проводящим частям с другой.
Перечень предприятий и цехов, попадающих в данную категорию очень большой. Практически все предприятия за исключением особо опасных входят в эту категорию.
Обязательно проведение мероприятий по охране труда и технике безопасности. По специальностям связанным с работой на вредном и опасном производстве проводится дополнительное обучение с аттестацией и допуском работников. Проводится аттестация рабочих мест.



На предприятиях в обязательном порядке проводится электротехническая экспертиза помещения по электробезопасности. На основании выводов экспертизы присваивается категория и на входе вывешивается специальный знак (табличка), на котором прописан класс помещения по электробезопасности.
К обслуживанию допускаются только квалифицированные специалисты прошедшие обучение и имеющие группу допуска в соответствии с требованиями охраны труда при обслуживании электроустановок.
3 категория: особо опасные помещения по электробезопасности
К особо опасным по электробезопасности помещениям относятся те, в которых имеется хотя бы один из приведённых ниже факторов:
- Особо сырые. Влажность воздуха 100%. Стены и оборудование покрываются влагой выпадающей в виде конденсата.
- Помещения с активной химической или органической средой, возникающей в помещении в течение рабочей смены. Эта среда разрушает детали электроустановок и изоляцию проводов.
- Если возникают одновременно два фактора относящихся к условиям повышенной опасности.



Эта категория помещений по электробезопасности имеет особые требования к используемому оборудованию и материалам. Предусматриваются более частые ТО и ремонты. Работает только квалифицированный и обученный к работе в определённых условиях персонал. Охрана труда, как правило, относит такие производства к вредным.
4 категория: территории, на которых установлены открытые электроустановки
К категории особо опасных относятся ОРУ — открытые распределительные устройства. Трансформаторные подстанции, распределительные узлы состоящие из огромного количества электрооборудования. Расположенных на открытой местности и огороженные забором. Это закрытые для несанкционированного проникновения территории, на которых действуют особые отраслевые требования по охране труда и квалификации работников.
Все помещения, аттестованные по электробезопасности должны обозначаться табличками, информирующими работников и представителей контролирующих органов о категории опасности за дверями.

Заключение
Установление классности помещений по электробезопасности процедура обязательная, но сама по себе ничего не меняющая. Статистика получения электротравм и несчастных случаев говорит о том, что это результат не столько слабых знаний ПУЭ и требований охраны труда, сколько неисправности электроустановок. Их несоответствия ПУЭ.
Помимо рисков электротравм следует иметь в виду и то, что электроустановки очень часто становятся причиной возникновения пожаров. Внимание МЧС к электрооборудованию и сетям, всегда независимо от категории помещений, повышенное.
Серьёзно к проверкам предприятий относятся СЭС и Роспотребнадзор. Эти органы интересуют условия труда работников. И они не пройдут мимо помещений с электроустановками.
Особое внимание государства говорит о серьёзности проблем в этой области. Следует ожидать ужесточение требований и ответственности. Появления новых нормативов и правил.
Видео по теме
Хорошая реклама
Чистые помещения для фармацевтических производств
Чистые помещения (или, так называемые, чистые комнаты) основательно вошли в фармацевтическую отрасль, и этому ненужно удивляться, ведь без них невозможно производство не одного лекарственного средства.
Чистое помещение (чистые комнаты или зоны) — это своего рода «помещение-барьер», которое служит препятствием для проникновения всевозможных контаминантов, а в воздухе, такого помещения поддерживается определённое количество частиц в определенном размере на один кубический метр. Такими частицами-контаминантами могут быть микроорганизмы, химические пары, аэрозольные частицы, частицы пыли или грязи.
Следует отметить, что проведения измерений счетной концентрации частиц в воздухе в чистых комнатах недостаточно, поэтому наряду с этим, подлежат периодическому мониторингу и такие параметры как, температура, давление и влажность. Чистые помещения, строятся и используются так, чтобы свести к минимуму поступление, генерацию и накопление таких частиц внутрь помещения.
Зачастую чистое помещение проектируется и строится внутри существующих зданий по принципу «здание в здании».
Для отделки чистых помещений обычно используют сэндвич-панели, и для стен, и для потолка. Сэндвич-панель — это панель, которая состоит из двух металлических листов с двухсторонним полимерным покрытием и наполнителем между листами, чаще всего, из минеральной базальтовой ваты или пенополиуретаном. Сэндвич-панели соединяются между собой особенными замками специального назначения с жесткой фиксацией, а все стыки подлежат герметизации. Стеновые панели крепятся на алюминиевый профиль, который монтируется на выровненный пол с применением фасонных скругляющих элементов, чтобы обеспечивать герметичность конструкций и удобство уборки чистых помещений. Сэндвич-панели хорошо выдерживают регулярную санитарную обработку помещений с применением всевозможных дезинфектантов. В запотолочном пространстве чистого помещения прокладываются инженерные коммуникации, например воздуховоды приточно-вытяжной вентиляции.
Пол чистой комнаты, может быть выполнен из специального сварного антистатического не пористого линолеума, который должен быть стойким к износу и легко поддаваться санитарным манипуляциям. Также полы чистого помещения могут быть наливными без сварных швов, что позволяет проводить эффективную очистку и дезинфекцию, которая необходима для подготовки чистого помещения к производству лекарственных средств (критически принципиально для лекарств, которые производятся в асептических условиях).
Необходимость в чистых помещениях и чистых зонах объясняется тем, что расположение производственного участка в условиях городской среды или промышленной зоны неизбежно приведут к загрязнению лекарственных средств совокупностью контаминантов из окружающей среды, если не выполнять фильтрацию воздуха с использованием высокоэффективных фильтров (HEPA).
Работающий персонал, технологическое оборудование и строительные конструкции генерируют загрязнения. В чистом помещении примерно 70-80% микро загрязнений приходятся на человека, 15-20%- на оборудование, 5-10%- на окружающую среду.
Чистота воздуха является критическим условием в производстве лекарственных средств, особенно стерильных лекарственных средств, и средств, производимых в асептических условиях, где нужны чистые помещения и чистые зоны, требуется обеспечения перепадов давления, микробиологической чистоты воздуха, чтобы производимые лекарственные средства не содержали патогенных микроорганизмов.
Для производства стерильных лекарственных средств правила GMP (Надлежащая производственная практика) устанавливают:
- классификацию чистых зон, включая предельно допустимые концентрации частиц в воздухе;
- значения скорости однонаправленного потока воздуха;
- величины перепадов давления между чистыми помещениями с различными классами чистоты;
- требования к контролю чистоты (мониторингу) и испытаниям чистых помещений;
- предельно допустимые концентрации микроорганизмов для разных зон;
- требования к персоналу, помещениям, инженерным системам.
Требования к концентрации частиц в воздухе различают для оснащенного и эксплуатирующего состояния.
В соответствии с действующим руководством по Надлежащей производственной практике:
Оснащенное состояние чистого помещения (at-rest) – состояние, в котором построенное и функционирующее чистое помещение укомплектовано оборудованием, которое полностью установлено, но технологический процесс не выполняется, а материалы, продукт и персонал отсутствует.
Эксплуатируемое состояние чистого помещения ( operational ) – состояние, в котором чистое помещения, функционирует установленным образом, с установленным технологическим оборудованием, с установленной численностью персонала, работающего в соответствии с документацией.
Измерение счетной концентрации частиц в воздушной среде чистых производственных помещений, производится оптическими счетчиками аэрозольных частиц, как в оснащенном, так и в эксплуатируемом состояниях чистых помещений.
Важной характеристикой чистых производственных помещений есть класс чистого помещения.
В таблице №1 представлена классификация чистых производственных зон, согласно требованиям GMP ЕС. Следует отметить, что стандарт GMP не является догмой, поэтому с учетом постоянного накопления нового опыта в сфере обращения лекарственных средств, требования к чистоте воздуха неоднократно менялись, но с 2008 года они неизменны.
Таблица №1
|
Зона |
Максимальное допустимое число частиц в 1 м³ воздуха, |
|||
|
В оснащенном состоянии |
В эксплуатируемом состоянии |
|||
|
0,5 мкм |
5,0 мкм |
0,5 мкм |
5,0 мкм |
|
|
А |
3 520 |
20 |
3 520 |
20 |
|
B |
3 520 |
29 |
352 000 |
2 900 |
|
C |
352 000 |
2 900 |
3 520 000 |
29 000 |
|
D |
3 520 000 |
29 000 |
Не регламентируется |
Не регламентируется |
Зона А — локальная зона для проведения критических операций в асептических условиях, где есть высокий риск для качества продукции. Важным моментом, в таких зонах есть однонаправленный поток воздуха, который обеспечивает в незамкнутой чистой зоне однородную скорость 0,36 -0,54 м/c.
Зона В — зона, которая окружает зону А и необходима для асептического производства;
Зоны С и D — чистые зоны, которые используются для менее ответственных стадий производства стерильной продукции.
Безусловно, на требования GMP к классификации чистых помещений, влияние оказал международный стандарт ISO 14664-1. Этот стандарт был принят в 1999 г. и установил единую классификацию чистоты воздуха по частицам для всех областей применений чистых помещений. В странах СНГ он был введен как ГОСТ ИСО 14664-1 «Чистые помещения и связанные с ними контролируемы среды. Часть 1 Классификация чистоты воздуха» в 2002 году. В Украине он был введен в 2004 году как ДСТУ ГОСТ ИСО 14664-1:2004.
На данный момент в правилах GMP ЕС, а также гармонизированных правилах GMP действующих в России и Украине к обозначениям зон A, B, C и D используется классификация по стандарту EN ISO 14644-1, который устанавливает классы чистоты, например,
- Класс А по количеству частиц в воздухе размером ≥5,0 мкм соответствует класс 4,8 ИСО, по частицам с размерам ≥0,5 мкм — класс 5 ИСО,
- Зона В в оснащённом состоянии соответствует класс 5 ИСО, а в эксплуатируемом – класс 7 ИСО для обоих значений пороговых частиц;
- Зона С соответствует классу чистоты 7 ИСО и 8 ИСО.
- Зона D в оснащённом состоянии соответствует классу чистоты 8 ИСО, а для эксплуатируемого состояния требования к чистоте воздуха по частицам не регламентируются.
Также следующим критическим фактором, который влияет на чистоту помещения для производства лекарственных средств, есть перепад давления. Он выступает своего рода барьером от различных загрязнений и от проникновения их из одного помещения в другое. Существуют, по крайней мере, три способа защиты от загрязнений:
- Защита процесса от окружающей среды;
- Защита окружающей среды от процесса;
- Одновременная защита процесса и окружающей среды друг от друга.
Эти способы, могут быть реализованы за счет, например перепадов давления между помещениями. Перепады давления между соседними помещениями разных классов чистоты должны быть 10-15 Па.
Если используются изоляторы, то перепад давления в изоляторе, должен быть более высоким по сравнению с обычными чистыми помещениями. При защите окружающей среды от процесса, где ведется работа с вредными веществами, давление должно быть ниже, чем в соседних помещениях. В случае необходимости защиты процесса и окружающей среды друг от друга, нужно предусмотреть чередование помещений с пониженными и повышенными давлениями воздуха.
При выше изложенных требованиях также следует проводить аттестацию (квалификацию) чистых помещений и текущий мониторинг, для подтверждения того, что чистые помещения соответствуют заданным требованиям.
Марина Титова
Менеджер Департамента Развития и Технической поддержки
Чистые помещение — проектирование и строительство, проекты «под ключ»
АТТЕСТАЦИЯ ЧИСТЫХ ПОМЕЩЕНИЙ
Чистое помещение – это инновационное техническое помещение, в котором концентрация частиц загрязняющих веществ поддерживается в определенных пределах в соответствии с требованиями стандартов производства различных продуктов.
Это помещение, в котором контролируется счетная концентрация аэрозольных частиц, построенное и используемое так, чтобы свести к минимуму поступление, генерацию и накопление частиц внутри помещения, и позволяющее при этом контролировать другие параметры, такие как температура, влажность и давление.
В основе архитектурно — строительных решений при создании чистого помещения лежит принцип построения «комната в комнате». Рабочая зона чистого помещения образуется пространством, ограниченным с помощью герметизированных элементов ограждающих конструкций.
Технология чистых помещений позволяет поддерживать чистоту воздуха на определенном уровне, который в свою очередь зависит от требований технологических процессов, проходящих в чистых комнатах.
В условиях фармацевтического, атомного, космического, биологического, оптического производств необходимы целые комплексы чистых помещений. В них включены как сами чистые комнаты, так и относящиеся к ним системы подготовки и очистки воздуха. Объем всего проекта обусловлен не только параметрами самого здания, но также требованиями технологических процессов и строительными нормами. Именно поэтому все этапы проектирования и строительства чистых помещений стандартизированы и описаны в ГОСТах.
Согласно государственному стандарту РФ «Чистые помещения и связанные с ними контролируемые среды» — ГОСТ Р ИСО 14644-4-2002, Часть 4. Проектирование, строительство и ввод в эксплуатацию», перед строительством должен быть разработан детальный план чистых помещений. Далее, после подготовки всех необходимых документов идет этап проектирования, где создаются чертежи будущих конструкций чистых помещений.
Перед вводом чистого помещения в эксплуатацию важно провести испытания. Целью проведения испытаний комплекса чистых помещений является проверка соответствия параметров чистых помещений в оснащённом состоянии (operational qualification (OQ)) заявленным классам чистоты по ГОСТ Р ИСО 14644-1-2002 «Чистые помещения и связанные с ними контролируемые среды», Часть 1, Классификация чистоты воздуха.
Если подтверждается, что чистое помещение функционирует согласно всем нормам и требованиям, а оборудование соответствует требованиям промышленной, пожарной и экологической безопасности, то помещение вводится в эксплуатацию. Аттестация чистого помещения — это процедура проверки режимов работы чистого помещения, документального подтверждения соответствия этих режимов нормам и правилам, мониторинг фактического значения счетной концентрации частиц в воздухе, а также других параметров установленных стандартом.
Состояния чистых помещений
- Построенное (as built): Помещение полностью смонтировано. Инженерные системы подключены. Оборудование, материалы и персонал — отсутствуют.
- Оснащённое (at rest): Помещение полностью укомплектовано работающим оборудованием. Персонал — отсутствует.
- Функционирующее (operational): Помещение функционирует установленным образом. Персонал — выполняет рабочие функции
ФГУП «ВНИИФТРИ» осуществляет комплексное испытание чистых помещений на стадиях DQ, IQ, OQ, PQ.
- Аттестация проекта (Design Qualification, DQ)— экспертиза, направленная на подтверждение корректности выполнения проектной документации.
- Аттестация в построенном состоянии (Installation Qualification, IQ)— документальное подтверждение полного соответствия смонтированного чистого помещения проектной документации.
- Аттестация в оснащенном состоянии (Operational Qualification, OQ)— документальное подтверждение соответствия всех строительных и инженерных систем утверждённому проекту. Заключение о правильном функционировании установленного оборудования.
- Аттестация в эксплуатируемом состоянии (Performance Qualification, PQ)— документальное подтверждение нормальных режимов работы всех систем. Полное соответствие требуемому классу чистоты, а также общим заявленным технологическим параметрам.
Основные операции, выполняемые во время испытания чистого помещения:
- Проверка объёма подаваемого и удаляемого воздуха
- Контроль движения воздуха между участками (зонами)
- Контроль правильности установки высокоэффективных фильтров
- Контроль течей воздуха через ограждающие конструкции
- Контроль движения воздушных потоков внутри чистого помещения
- Концентрация аэрозолей и микроорганизмов (если это необходимо)
- Дополнительные тесты
Квалификация склада | Центр Валидации
Существует ряд международных требований (GMP, GDP, GSP) в которых сказано, что квалификация склада (фармацевтического, аптечного, дистрибьюторского) должна быть проведена. Этот процесс необходим для того, чтобы убедиться, что условия хранения лекарственных средств и материалов соответствуют указанной информации на упаковке, и учитывают температуру и относительную влажность воздуха.
Квалификация склада — это процесс документального подтверждения того, что условия хранения продукции соблюдаются по всей площади и объему склада, на каждой полке, в местах хранения продукции.
Система вентиляции, кондиционирования и обогрева воздуха фармацевтического склада должна поддерживать заданные параметры температуры и, при необходимости, влажности воздуха независимо от времени суток, дня недели или климатического сезона. Соответственно, квалификация склада должна охватывать эти условия и проводиться, как минимум, для двух наиболее критических климатических сезонов, когда температура окружающей среды выше и ниже, чем температура хранения продукции в складе, что соответствует работе систем охлаждения в «теплый» период года и систем обогрева в «холодный» период года.
Процесс проведения
Первым этапом квалификации склада может быть квалификация проекта. Она проводится до начала строительства и заключается в анализе проектной документации. Квалификация проекта не является обязательной, но именно на этой стадии могут быть выявлены критические ошибки, исправить которые после реализации проекта будет не просто, если вообще возможно.
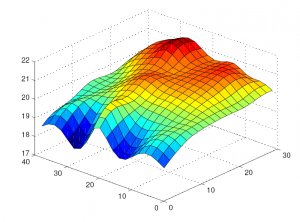 Квалификация монтажа проводится для новых складов или после их реконструкции. На этой стадии фармацевтического склада в основном используется методы визуального контроля и сравнения со спецификацией. Проводится контроль наличия проектной документации, инструкций по эксплуатации и обслуживания элементов системы вентиляции, воздухоохладителей, нагревателей, систем автоматизации и диспетчеризации и др. Также необходимо проверить монтаж помещений, оборудования, трубопроводов и вспомогательных систем на соответствие действующим техническим чертежам и спецификациям.
Квалификация монтажа проводится для новых складов или после их реконструкции. На этой стадии фармацевтического склада в основном используется методы визуального контроля и сравнения со спецификацией. Проводится контроль наличия проектной документации, инструкций по эксплуатации и обслуживания элементов системы вентиляции, воздухоохладителей, нагревателей, систем автоматизации и диспетчеризации и др. Также необходимо проверить монтаж помещений, оборудования, трубопроводов и вспомогательных систем на соответствие действующим техническим чертежам и спецификациям.
В соответствии требованиями GDP на стадии квалификации монтажа необходимо определить требования к калибровке оборудования систем мониторинга и проверить наличие сертификатов калибровки. Мы рекомендуем разделять процесс квалификации склада и монтаж системы мониторинга, т. к. он должен проводиться после квалификации склада и определения критических точек контроля. Не следует забывать, что система мониторинга также нуждается в квалификации, а это уже совсем другой процесс.
Квалификация функционирования проводится после успешного завершения квалификации монтажа. На этой стадии осуществляется картирование склада с пользованием беспроводных даталоггеров температуры и влажности. Применение тепловизоров для этого процесса некорректно, ИК-термометры можно рассматривать лишь как дополнительный инструмент анализа рисков. По своей сути картирование склада (маппинг склада) — это определение наиболее холодной и горячей точки, а также точки в которой наблюдается максимальное колебание температуры. Эти измерения проводится на протяжении минимум 24 часов с интервалом измерений 5-15 мин, количество даталоггеров определяется индивидуально для каждого помещения хранения.
По результатам выполненных измерений склада должны быть определены критические точки контроля для пустого склада, в которые должны быть размещены датчики системы мониторинга.
После успешного завершения квалификации функционирования должны быть оформлены письменные методики (СРМ, СОП), должны быть завершены все работы не связанные с дистрибьюторской деятельностью, закончено проведение обучения операторов. Все это позволит использовать фармацевтический склад.
Квалификация эксплуатации проводится после успешного завершения стадий квалификации монтажа и функционирования. На этой стадии производится картирование склада, аналогично выполненному на стадии квалификации функционирования, только на складе загруженном продукцией. Измерения проводится на протяжении минимум 72 часов. Объем заполнения склада должен быть максимально возможным, для моделирования наихудшего случая с точки зрения препятствия распределения воздушным потокам, обычно это более 60%.
По результатам выполненных измерений склада должны быть определены критические точки контроля для склада с продукцией, в которые, если они отличаются от определенных на стадии квалификации функционирования, должны быть размещены дополнительные датчики системы мониторинга.
Стадию квалификации эксплуатации необходимо проводить минимум для «холодного» и «теплого» периодов года для подтверждения работы системы обогрева и охлаждения. В случае если система вентиляции, кондиционирования и отопления оснащена системой автоматического перехода тепло↔холод, необходимо проведение квалификации 3 раза в год:
-
В холодный период — зимой, когда среднесуточная температура окружающей среды ниже температуры хранения;
-
В теплый период — летом, когда среднесуточная температура окружающей среды выше температуры хранения;
-
Переходной период — осень/зима или зима/весна, когда температура ночью опускается ниже температуры хранения, а днем — выше температуры хранения.
Повторная квалификация склада
GDP говорит о том, что периодичность квалификации необходимо основывать на анализе рисков, контроле изменений и обзорах данных системы мониторинга. Однако, как показывает практика анализ рисков проводится формально, с заведомо известным результатом, преследующим цель «ничего не изменено», система контроля изменений функционирует, в лучшем случае, по накопительной системе, об управлении изменениями речи не идет, а обзоры данных системы мониторинга проводятся без оценки тенденций, а лишь по принципу «входит в пределы». Исходя из этого, в большинстве случаев, периодичность квалификации 1 раз в 2, 3 или более лет является необоснованной, а при проведении квалификации на таких складах выявляется большое количество проблем.
Лучшей практикой планирования и организации работ по квалификации дистрибьютора лекарственных средств является установление периодичности реквалификации, которая охватывала бы теплый и холодный периоды года, на протяжении каждого года, для наработки достаточной статистики, которая и позволила бы снизить периодичность реквалификации.
Условия складского хранения должны быть безопасными и надежными независимо от времени суток, дня недели или климатического сезона.